Parthasarathy Ganguly
Physical Chemistry Division
National Chemical Laboratory
Homi-Bhabha Road, Pashan
Pune 411008, INDIA
Abstract.
Principles of molecular tensegrity have been developed earlier by treating molecules as tensegrity structures and applied to gas-phase MXn molecules. These principles are now applied to A…H-B hydrogen bond complexes. In this approach a tensegrity factor is obtained from the ratio of ideal A…H or H-B distances to that of ideal A…B distances. These ideal distances are obtained from sizes, CR0, of charged species of A, H, and B atoms obtained from their atomic size descriptor, rnZc. Limiting values of various A…B distances for “n-polar” (ionic) and “neutral” sizes that correspond to strong and weak hydrogen bonds are obtained without requiring a knowledge of the hydrogen atom positions. These distances are then compared with (> 1000) observed A…B distances in compounds of biologically important amino acids. O…H-O and O…H-N hydrogen bonds are strong or “n-polar” in character in this analysis for a given coordination number. The very short hydrogen bonds (SSHBs or LBHBs) are found to those with a large coordination numbercontacts which are shorter than “neutral” or van der Waals contacts. The way the molecular tensegrity of the A…H-B hydrogen bond complexes impacts the length of A…H and H-B bonds are discussed.
1. Introduction
The van der Waals’ sizes, rvdW, of atoms have been used1/36 to characterize hydrogen bond distances, even if there has been no unambiguous theoretical inputs into the way these sizes can be obtained from a balance of exchange repulsion and attractive dispersion forces that is thought to characterize a van der Waals interaction. There is also a fickle-ness involved in identifying the actual pair of atoms in the A…H-B complex which are to be evaluated by their interatomic separations. It has been the conventional wisdom that the proton in the hydrogen bond is buried in the electron cloud of the B atom in an A…H-B linkage such that the hydrogen bond contacts are just the sum of van der Waals’ radii of A and B atoms. The formation of a hydrogen bond is also usually indicated when the A…H distance is smaller than the sum of the van der Waals’ radii of the A and H atom. Such a shortening of the distance has been conventionally attributed2/2 to the formation of three-centre four-electron bonds with considerable electron overlap between the orbitals of A, H and B atoms. The discrete assumption is that despite such overlap, the predominant interaction is electrostatic so as to account for flexibility in bond lengths and AHB bond angles.
It is pertinent to note from a historical point of view that in the first description of hydrogen bonding Latimer and Rodebush3, 4/52, 53 stressed the importance of the high dielectric constants of hydrogen bonded systems. This has been attributed to proton displacements from the centre of the atom. An important consequence of such atomic displacements in Latimer’s model or as a consequence of asymmetric bonding effects in modern theoretical approaches is that the spherical atom approximation is no longer valid. This poses considerable difficulty5/54 in locating the hydrogen atom from electron densities obtained from X-ray diffraction studies since the hydrogen has no core electron to identify it by. On the other hand, since electron densities are to be related to chemical reactivity, one may be actually interested in the electron density profiles obtained by X-ray refinements. In neutron diffraction studies the position of the nucleus is more accurately determined as the nuclear position may be accurately represented as a point-scatterer. The discrepancies between X-ray and neutron diffraction results affect the way the non-bonded A…H distance is to be evaluated. This ambiguity in locating the hydrogen atom is inevitable and unavoidable. One may escape from this dilemma if the A…B distance in the A…H-B complex provides a measure of the contribution of the H atom to the interatomic distances without actually requiring an accurate estimate of the position of the hydrogen atom.
In this submission we examine the values of expected A…B distance in A…H-B hydrogen bond complexes from considerations of molecular tensegrity. This concept has been used earlier6-8/40, 41 for obtaining 1,3- non-bonded distance in X-M-X’ linkages in gas-phase MXn compounds. In this approach (see section 2.3), the ideal 1,3-X…X’ distances are obtained from a knowledge of “ideal” “charge-transfer” single-bond M-X distances, d00, and a size, CR0–(X), of the X atom, which are themselves simple linear function9/38 of atom-specific sizes9-11/37-39, rnZc. There is no requirement for knowing the actual M-X or M-X’ distances. We apply these concepts to the A…H-B complex in a novel manner using simple mechanical concepts which treat molecules as tensegrity structures and which have been used earlier to obtain quantitative information on 1,3- distances. We only assume that there are “ideal” non-bonded A…B and bonded H-B distances that allows a tensegrity factor to be defined and the A…B distance calculated given the atomic sizes of the A and B atoms and without knowing the actual location of the hydrogen atom.
There are other debates12-14/3, 5, 6 on the way the interatomic A…B van der Waals contact distances should be compared by taking into account the AHB bond angle. The van der Waals dispersion reaction interaction between atoms is a consequence mainly of the induced-dipole induced-dipole interactions between atoms which is isotropic, to first order. As such, the A…B van der Waals interaction in the A…H-B hydrogen bond complex will not be determined by the AHB angle. We emphasize, in particular, the way the “ionic” (or “n-polar”) sizes9, 10, CR–, and the “neutral” (or vdW) sizes, rvdW, may be used to obtain cut-off limits for the A…B distances in strong and weak hydrogen bonds in A…H-B complexes.
Another deficiency in discussing hydrogen bonds in terms of atomic sizes is that there is no discussion on the way very short hydrogen bonds may be accommodated within the framework of atomic sizes that have been normally used. This aspect is important since there has been renewed interest13, 14/5, 6 in hydrogen bonding because of the possibility for low-barrier hydrogen bonds (LBHBs) playing an important role15-17/8, 9, 18 in important biological processes. The formation of a LBHB is useful in stabilizing the transition state and enhancing the reaction rate in acid-base catalyzed reactions15-17/8, 9, 18. The very short A…B length (e.g., ~ 240 pm in LBHB O…H-O complexes as compared to ~ 280 pm in normal O…H-O hydrogen bonds) in a A…H-B hydrogen bond complex is taken as a measure of the existence of LBHBs which are also sometimes identified as short strong hydrogen bonds (SSHBs). We find that we may use dependence of the tensegrity factor6-8/40-42 on local coordination number provides possible insights into the shortening of 1,3- distances including those in A…H-B complexes.
2. Atomic Sizes, Interatomic Distances and Molecular Tensegrity
2.1. Atomic Sizes.
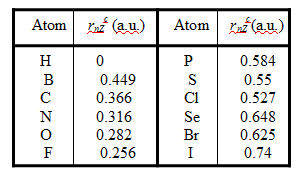
The atomic sizes, rnZc, used by us have been obtained from a classical stationary point in a new model9-11/37-39 and without adjustable parameters. The basic premise of this model is that atomic sizes are defined by external interaction which is represented by the absorption or emission of a photonor a virtual photon in vacuumwhich, in turn, is represented by an electron-hole pair, (e–-h+). The atomic size is obtained9-11/37-39 by considering the interaction of the outer-electrons with the hole, h+, of the electron-hole pair that represents the external interaction. Because h+ is an universal component of the external interaction field, (e—h+), the atomic size thus obtained is not dependent on the actual nature of the external interaction. It is, however, atom-specific, because it is dependent on the way the outer electrons are distributed, say, between the valence and inner shells, as well as the way the d- and f- electrons of transition metal elements are treated. This is the new paradigm shift in which an external field is used to define an atomic size instead of calculating the size of an isolated atom internally. Some of the sizes of atoms that we will be using in this submission are given in Table 1 (note that the sizes are in atomic units).
Table 1. Atomic Sizes for some atoms relevant for this work.
2.2. 1,2-Interatomic Bonded Distances.
A general expression9/38 for interatomic distances, dM-X, for an M-X bond is written (for convenience) in terms of a “hub” and “axle” description with
dMX(cal) = eff[{CMrnZc(M)/FS(M) + CXrnZc(X)/FS(X)}”hub”
+ {DM/FS(M) + DX/FS(X)}“axle”] (1)
The term FS takes18, 19/48, 49 into account the shortening of bond distances due to the presence of nv “unsaturated” (or what we henceforth term as “extrabonding”) electrons. FS is empirically found18, 19 to be FS = 1.18, 1.26, 1.32, 1.38 and 1.42 for nv = 1, 2, 3, 4 and 5, respectively. Writing nv in terms of a spin Sv (= nv/2) valence electrons that contribute20/55 to bond order (= nv + 1) we write FS [1 + (2/)2{Sv(Sv+1)}1/3. For most part of this article we will require FS = 1.
The values of the coefficients CM,X or DM,X for the “hub” and “axle” sizes for M and X atoms usually correspond to either “charge-transfer” values (C+ = 2.144, C– = 2.300, D+ = -2aH/3, D– = 2aH) or “neutral” values (C = 1, 2, …, D = 1, 2 …). In what follows we will require an “ideal” (eff = 1) “single bond” (FS = 1) “charge-transfer” distance, d00, for the non-transition metal elements we study in this paper. Thus the ideal M-X (rnZc(M) rnZc(X)) single bond charge-transfer distance is given by9/38 (in pm)
dMX00 CR0+(M) + CR0–(X) (2a)
CR0 C0rnZc + D0 (2b)
with C0+ = 2.144, C0– = 2.30, D0+ = -2aH/3 and D0– = 2aH. We thus obtain
dMX00 2.144rnZc(M) + 2.30rnZc(X) + 4aH/3 (3)
The zeros in the superscript or subscript indicates that there are no “extrabonding” valence electrons or nv = 0.
2.3. Molecular Tensegrity and 1,3-Distances.
For an X-M-X linkage, we have used6-8 a “tensegrity factor”, t00, as a measure of the matching of idealized “charge-transfer” M-X (eqn 3) and X- – -X distances. The“ideal” charge-transfer separation, dXX00=, between the X atoms in the X-M-X linkage is obtained from eqn 2b as
dXX00= = 2CR– = 2(2.300rnZc(X) + 2aH) (4)
From these considerations, the tensegrity factor, tMX00 is obtained as
tMX 00 = dMX00 /dXX00= 0.5[CR0+(M)/CR0–(X) + 1] (5)
In the way eqns 1 – 4 are written, t00 is dependent only on the core atomic size, rnZc, of M and X atoms without requiring separate estimates of ionic character, for example. It is also dependent (eqn 5) on the ratio R = CR0+(M)/CR0–(X), which is the radius ratio of the charge-transfer sizes of M and X atoms. It is well known that there are geometrical limits to the value of R for various coorination numbers, N. For example, for N = 4 and N = 6 the upper limiting values of R are 0.414 and 0.732, respectively. One then obtaining values, t00limN for the limiting values of R. Thus, t00limN = 0.707 and 0.866 for N = 4 and 6, respectively.

Fig 1. Plots of observed X…X distances (from ref 21) in gas-phase MXn compounds (X and M are atoms of insulating elements) vs calculated distances from eqns 6-8 and atomic sizes from Table 1. Circles: n 4, N = 4, K = 1; Squares: N = 6, MF6 compounds (M = S, K = Kpolar = 1; M = Se, K = (Kpolar + Kneutral)/2 = 1.0625; M = Te, K = Kneutral = 1.125).
A term FS*N which given by
FS*N = [2 – t00/t00limN] (6)
is now introduced as a measure of the matching of the 1,2- M-X and the 1,3- X…X distances with FS*N = 1 when t00/t00limN = 1. When FS*N > 1 there is a contractive pressure on the 1,3- X…X distance tending to compress it from its ideal value, 2CR0–(X) (eqn 4). When FS*N < 1 the 1,3-distance would tend to expand over 2CR0–(X). The X…X distance for gas-phase MXn compound may be written in terms of a size CR(X) as
dX—X 1,3 = 2CR(X)1,3/FS*N (7a)
= 2KXX[2.3rnZc(X) + 2aH]/ [2 – t00/t00limN] (7b)
The size CR(X) = KXXCR0–(X). K = 1 or K = 1.125 corresponding10/39 to “n-polar” (or “ionic”) and “neutral” (or van der Waals) sizes of the X atom, respectively. The term, XX, is an effective dielectric constant which allows for small changes due to environmental influences in a manner consistent with the size of the atom. For the purpose of this communication we use the empirical relationship
XX = 1+ [0.0019{2(2.3rnZc + 2aH)}]6 (8)
For an X-M-Y linkage the X…Y distance has been found to be given by
dX—X’ 1,3 = {CR(X) + CR(Y)} /FS*N (9a)
= KXXX{2.3rnZc(X) + 2aH}/{2 – tMX00/tMX00limN} +
KYYY{2.3rnZc(Y) + 2aH}/{2 – tMY00/tMY00limN} (9b)
The provision is made that KX need not be the same as KY and could have as before K = Kpolar = 1.00 or K = Kneutral = 1.125 for “n-polar” or “neutral” distances.
For gas-phase MXn compounds (n 4) where M and X are atoms of insulating elements (except SiH4, GeH4 and KrF2 and XeF2) we find that K = 1 and N = 4, gives the 1,3-X…X distances satisfactorily as shown in Fig 1. For MX6 compounds (n = 6) the calculated values are too large when we use N = 4 in eqns 6 and 7, even with K = 1 = XX. Good fits are obtained only when we use N = 6 and allow for variations in K (see fig 1 for MF6 compounds; M = S, Se, Te). This highlights the way an increase in coordination number could lead to a decrease in the 1,3- distances. We shall use this aspect for understanding short hydrogen bond lengths15-17 in LBHBs (or SSHBs).
3. A…B Distances in Hydrogen-Bonded A…H-B complexes.
A point of detailed interest in this communication is to examine the A…B distance as a 1,3-distance in a A…H-B hydrogen bond in terms of the molecular tensegrity model borrowed from Buckminster Fuller’s analysis of engineering tensile integrity structures. For the purpose of this communication we have chosen hydrogen bond complexes in compounds of biologically important -amino acids. We have chosen all A…H-B complexes in which the A…H distance is less than the sum of their “neutral” sizes CR(X) = KCR0–(X) distances with K = 1.125. We have normalized the reported A…H distances, dA—H by dividing by of the sum of the “n-polar” (K = 1) sizes of A and H atoms to obtain a ration RA—H. The histograms of the ratio A—H obtained from the dA—H distances obtained from CCDC tables for O…H-O, O…H-N and other A…H-B complexes used in this study are shown in Fig 2. It is seen that in all these systems the A…H distances are less than the sum of their “neutral” sizes or RA—H < 1.125.
We treat the A…H-B complex as similar to the X-M-Y linkage and apply equations similar to those in eqn 9. The “ideal” non-bonded A…H distance in the “charge-transfer” model are taken as the sum of the “n-polar” sizes of A and H atoms. We then write (in pm)
dA- – -H00= = CR0–(A) + CR0–(H) CR0–(A) + D0–(H) (10)
= (2.30rnZc + 105.8) + 2aH (11)
The tensegrity factor, t00=, for the A…H component is then that of a hypothetical A…H…A linkage. The = sign in the superscript of eqn 10 is to indicate that the ideal A…H distance is the sum of “n-polar” sizes of the A and H atoms. The value of tA—H 00= is written as
tA- – -H00= = dA- – -H00=/2CR0–(A) (12)
The tensegrity factor tH-B00 is obtained in the usual way from eqn 5 as
tH-B00 = dB-H00/2CR0–(B) (13a)
= (2. 144rnZc(B) + 70.6)/(2.3rnZc(B) + 105.8) (in pm) (13b)
The A…B distance is then obtained in a manner similar to that from eqn 9 as
dA—B = KAAA{2.3rnZc(A) + 105.8}/{2 – tA—H00=/tA—H00=limN} +
KBBB{2.3rnZc(B) + 2aH}/{2 – tB-H00/tB-H00limN} (14)
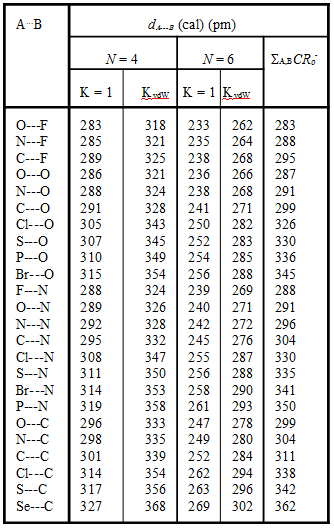
The A…B distance can then be calculated (Table 2) from eqn 14 using the atomic sizes, rnZc some of which are given in Table 1, for a given value of K and values of XX from eqn 8. The last column in Table 2 gives the sum of the charge-transfer “n-polar” size, CR0– (eqn 2b) of the A and B atoms. For small atomic sizes (e.g., F, O, N) the value of A,BCR0– is close to that expected from the tensegrity model with N = 4 and K = 1. The calculated N = 4, K = 1 distances of O…F, N…F, C…F and C…C are to be compared with the distances of 266, 280, 305 and 367 pm, respectively, used by Buckingham and Fowler in their classic paper.1/36.
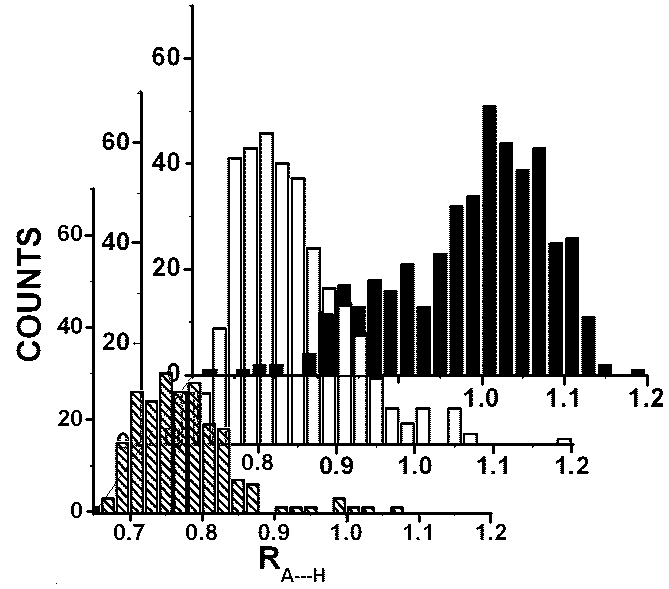
Fig 2. Histograms of the ratio RA—H of observed A…H distances, dA—H, and the calculated “n-polar” size [CR0–(A) + CR0–(H)]. Hatched boxes: O…H-O; Open boxes: O…H-N; filled boxes: other A…H-B complexes.
The calculated distances have been compared with those observed in these compounds of biologically important amino acids by normalizing the observed A…B distances with that calculated from eqn 14 with N = 4 and K = 1 and AA or BB calculated from eqn 8. The resulting ratio of observed and calculated distances is termed as RA—B. We show in Fig 3 the histograms of the values of RA—B of the distances observed using as a upper cut-off the distances given as the sum of van der Waals sizes in the CCDC tables. We see that there are two maxima around R = 1 and R = 1.125. The larger contributions may be attributed to “n-polar” (K = 1) sizes while the value of RA—B for the other maximum is close to that are to be attributed to “neutral” or vdW (K = 1.125) sizes.
In order to look at more specific details we have shown in Fig 4 the corresponding histograms of the O…O, O…N and O…C distances. As expected, a comparisons of figs 3 and 43 shows that the predominant contribution to the histogram in fig 3 is from the O…N linkages. The maximum in the histogram for RO—N is at values marginally less than 1. This may be attributed to rather large calculated (eqn 8) values of OO (~ 1.023) and NN (~ 1.027). The maximum in the histogram for RO—N is at values greater than 1 when OO = NN = 1 is used. The RO—O histogram shows again a bimodal distribution. The values of RO—O are considerably less than 1. The maximum in RO—O slightly less than 1 even when OO = 1. The second maximum is near the average of the values calculated for RO—O (~ 0.91) from N = 4 and N = 6 and K = 1. The values of RO—O calculated with N = 6 and K = 1.125 (“neutral” sizes) is close to 0.93. It is apparent that the short O…O distances may be related to higher coordination number around the O…H-O complex. As an example, we have shown in fig 5 the number of contact distance which are less than the sum of van der Waals radii as per CCDC for compounds with short (~ 240 pm L-alanine alaninium nitrate) and another with normal (~ 280 pm cis-4-hydroxypiperidinone) O…O distance. The former has a larger number of contacts which would be consistent with a higher value of N.

Fig 3. Histograms of the ratio RA—B of observed A…B distance in crystals of various compounds of amino acids as compiled in the literature and the calculated A…B distance using eqn 14 with K = 1 and rnZc taken from Table 1. The vertical lines indicate the values calculated for the given values of K with the value of N given in brackets. The meaning of <4,6> is that it is the arithmetic average of values expected for N = 4 and N = 6.
When N = 6 and K = 1 in eqn 14 RA—B ~ 0.82. For the O—O linkage the expected distance is nearly 236 pm (Table 2). A very short separation between carboxylate oxygens of only 230 pm has been claimed22/17 for the active site of HIV-1 protease (PR). Usually a A…B distance of ~ 280 pm (K =1, N =14 in Table 2) is an indication of a normal hydrogen bond distance in O…H-O or O…H-N complexes with an enthalpy of formation close to -5 kcal/mol. Such hydrogen bonds are asymmetric as far as the location of the proton between the H and B atoms are concerned. Asymmetric proton locations persist even when O…O distances are as small as 250-260 pm. It is sometimes thought that A…B distances close to 240 pm could signal the existence of a single well hydrogen bond with the proton located symmetrically between the A and B atoms.
Table 2. Values of A…B distances (in pm) in A…H-B hydrogen bond complexes calculated from eqns 1-8 using various values of K and N and rnZc from Table 1.
The histogram for RO…C in these compounds of amino acids shows that RO—C > 1 with a maximum near K = 1.125. This would be consistent with the O…C linkage being described as one involving “neutral” contacts (or more proximate to van der Waals contacts) in the hydrogen bond O…H-O complex. The RA—B histograms for A…B linkages other than those shown in Fig 4 are shown in Fig 6. Like the O…C linkages the histograms show mainly the populations of the RA—B > 1.00 values. The histograms of RC—O populations are shown by black boxes in the main diagram while the RC—C histograms are shown as inset in Fig 5. Among those with RA—B < 1 are C…O (0 out of 45), C…N (1 out of 44), C…C (1 out of 28), Cl…N (2 out of 21) or Cl…O (5 out of 21). Thus the A elements which are not N or O (fluorine has not been counted as there are not enough examples) usually have RA—B > 1 and sow a maximum in the distribution at values close to 1.125 (N = 4, K = 1.125 = Kneutral). We have shown the histogram for RC…C in the main part of fig 5.

Fig 4. Histograms of RO—O (hatched boxes), RO—N (open boxes) and RO—C (filled boxes). The plane of RA—B = 1 is shown as a guide to the eye. The scale is the same for all the three histograms.
The above analysis indicates that “n-polar” (K = 1 in eqn 14) hydrogen bonds with RA—B < 1 are likely only when B = O or N. When A = N or O and B N or O RA—B > 1 (for K = 1 in eqn 14) indicating that in this case “neutral” hydrogen bond complexes may be formed, which could be similar to van der Waals complexes. This aspect could be consistent with our model for interatomic distances. For example, in the H-B bond distance requires the size CR0+ for the B atom (eqn 2, rnZc (O or N) > rnZc(H) = 0, Table 1). The size CR0+ is negative for oxygen (and fluorine) and close to 0 for nitrogen. Even in the case of nitrogen the size CR0+(N) is negative (~ – 6 pm) when we use the empirical size, RG(N) = 0.26 a.u. and the relationship CR0– = 2.25rG – 37 (in pm). Such empirical relationships are useful as they pragmatically take into changes in environmental factors such as those that contribute to eff in eqn 1 especially when they are different for the
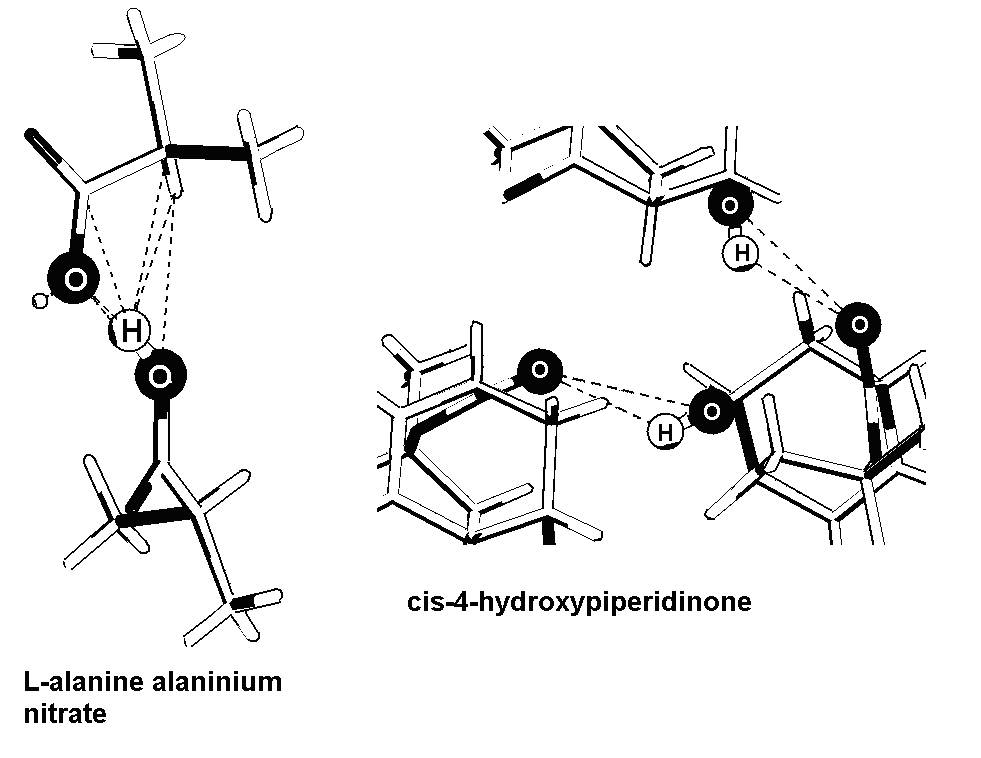
Fig 5. Showing the contact distances of the atoms in O…H-O hydrogen bonded complexes of L-alanine alaninium nitrate (from ref 23/60, dO—O ~ 240 pm) and cis-4-hydroxypiperidinone (~ 280 pm, from ref 24/59).
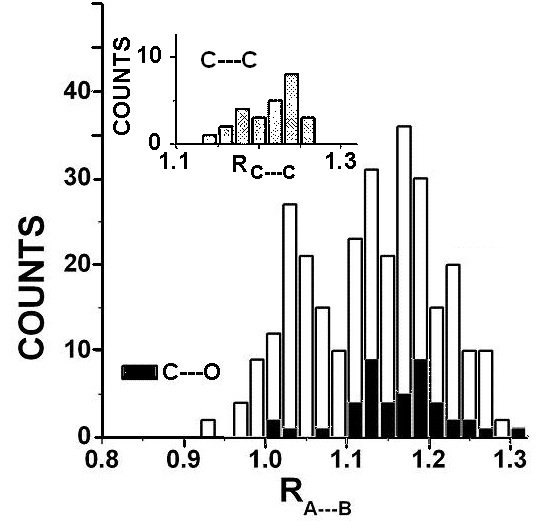
Fig 6. Histograms of RA—B (other than O…O, O…N and O…C linkages. The RC—O histograms have black boxes and the RC—C histogram is shown in the inset. hatched boxes), RO—N (open boxes) and RO—C (filled boxes). The plane of RA—B = 1 is shown as a guide to the eye. The scale is the same for all the three histograms.
“hub” and “axle” components. The nucleus of an atom with a negative value of CR+ is likely to be exposed to the valence electron of another atom25/61. The A atom in a Y’-A linkage is on the other hand more electronegative than the A atom (rnZc(Y’) > rnZcA)) so that one associates the size CR0–(A) or a negative charge with the A atom. Such complementary size-symbiosis in CR0–(A) and CR0+(B) when B = O, F and N would indicate the fundamental basis for the “n-polar” (K = 1) description for most A…B linkages when B = F, O, and N.
4. Systematic Changes in Interatomic Distance in A…H-B Hydrogen Bond Complexes.
Although the 1,3- A…B distance in A…H-B hydrogen bond complexes are amenable to analysis in the molecular tensegrity model using eqn 14, for example, it may be necessary to understand the way the A…H and H-B distances comply with changes dictated by the tensegrity of the A…H-B complex. After all, it is rather likely that the disposition of the hydrogen atoms would be most necessary in allocating the nature of biochemical reactivity to the hydrogen bond complex. We expect first of all a fairly linear relationship between the A…B distance, dA—B, and the A…H distance. dA—H. We find that the best linear relationship is observed (fig 6) between dA—B and the component, dA—HAB (= cos(HAB)dA—H) of dA—H in the A…B direction instead of the linear fit between dA—B and dA—H. The best linear fit is given by (R > 0.95, SD = 10 pm)
dA—B = 0.94dA—HAB + 98 (in pm) (15a)
while the best fit with unit slope is given by
dA—B = dA—HAB + 85 (in pm) (15b)
These fits are marginally better than the the fits between dA—B and dA—H (R > 0.93, SD = 12 pm) :-
dA—B = 0.90dA—HAB + 105 (in pm) (16a)
while the best fit with unit slope is given by
dA—B = dA—HAB + 84 (in pm) (16b)
The relationships in eqns 15 and 16 highlight the synergistic behavior expected for the geometry of teh A…H-B complexes.
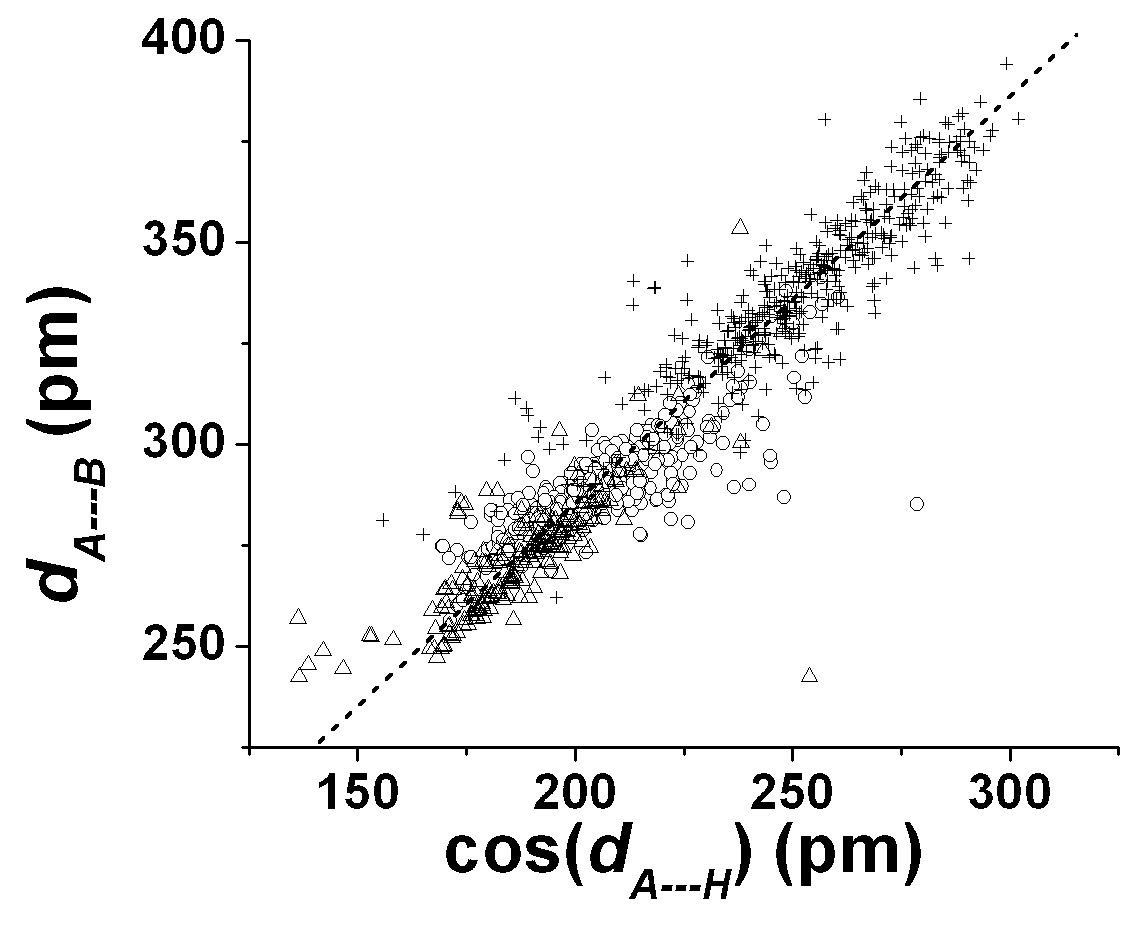
Fig 7. Plots of dA—B versus cos(dA—H) which is the component of dA—H along the A—B direction. Triangles: O…H-O bonds; circles: O—H-N bonds; + sign: other A…H-B bonds. Dashed line is meant as a guide to the eye for unit slope.
The H-B distance, dH-B, has been renormalized in terms of the calculated distance using eqns 1-4 (eff = 1) to give dH-Bnor = dH-B(2.144rnZc(B) = 2aH). Similarly, the normalized distance, dA—Hnor is obtained as dA—H nor = dA—H/(CR0–(X) + CR0–(H) using eqns 2-4. We thereby obtain the ratio RH-Bnor = dA—Hnor/dH-Bnor. We have plotted in Fig 7 the distribution of RH-Bnor for O…H-O, O…H-N and other A…H-B complexes and have compared them with the corresponding distribution of RA—B (= dA—B/dA—B(cal)) of Fig 3 (calculated from eqn 14 with N = 4, K = 1, and XX = 1). We have also shown the distribution of dH-Bnor and dA—Hnor for O…H-O hydrogen complexes. It is seen that the distribution of the ratio RH—Bnor agrees more with RA—B than either of the distributions dA—Hnor or dH-Bnor. The considerably larger width of the distribution RH—Bnor as compared to to RA—B is to vbe expected because it is the ratio of two distributions. The interesting feature is that it is the ratio RH—Bnor that agrees better with RA—B than the distributions dA—Hnor or dH-Bnor. This would indicate that dA—B has contributions directly related to dA—H and inversely related to dH-B. For a given dA—B one could then expect the dH-B distance to expand as dA—H is increased and vice versa. This mutual dependence of dH-B and dA—H has been one of the more important features of A…H-B hydrogen bond complexes.12/3
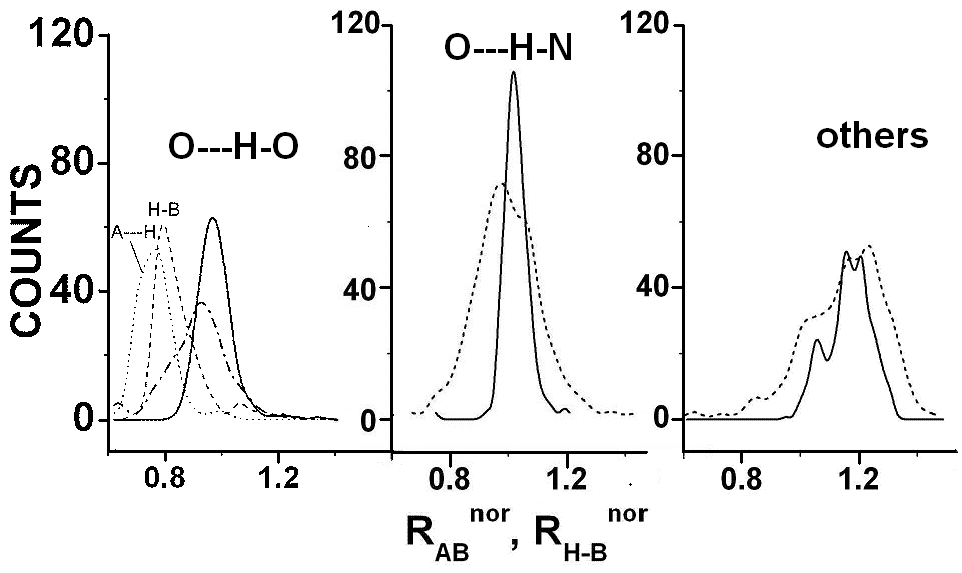
Fig 8. Plots of distribution of various bond distances in A…H-B complexes for (see text)
RA—Bnor (thick full lines) and RH—Bnor (thick dashed lines): O…H-O (left of diagram), O…H-N (centre) and others (right). The distribution of (see text) dA—Hnor and dH-Bnor for O…H-O complexes are also shown.
5. More on Short Hydrogen Bonds and LBHBs.
The hydrogen bond between homo-nuclear atoms is especially interesting. One may consider the equilibrium between two hydrogen-bonded states such as
Oa…H-Ob Oa-H…Ob (3)
The initial and final states may be considered to be degenerate as long as the environments of the two oxygen atoms, Oa and Ob, are identical. In chemical terms this would translate into the two oxygen atoms having pKa values close to each other. The important feature of eqn 3 is that the initial state is degenerate with the final state once there is no change in the nuclear position of the oxygen atoms relative to that of the hydrogen atom after the exchange. In this case there could be low-barrier “itinerant” hydrogen bonds with exchange of the H atoms between Oa and Ob atoms. There is likely to be pKa matching in this case and the proton could be itinerant between the two structures in eqn 3.
The critical feature of importance is the degeneracy of the initial and final states in eqn 3. This feature was first recognized in Zener double exchange26 systems, as visualized in the manganites27, for example. In mixed valence, Mm+-M(m-1)+ systems (m is the number of valence d electrons, say) the equivalent electron transfer reaction has been written as
Mam+-Mb(m-1)+ Ma (m-1)+…(e)-… Mb (m-1)+ Ma (m-1)+-Mb (m)+ (17)
When the initial state is degenerate with the final state in eqn 17, the energetically uppermost valence electron is itinerant between the atoms Ma and Mb. The individual valence states of the M atoms are not distinguishable (homogeneous mixed valence) when the measurement time is less than hopping frequency. In Zener double exchange systems, the spin of the electron plays an important role is plays an important part in introducing ferromagnetic coupling between M atoms when m > 1 as in the perovskite manganites27. This is because the electron-spin requires to be conserved during the exchange process as in eqn 5 below (m = 4; e.g., Mn3+-Mn4+ in manganites).
Ma-Mb Ma…(e–)-Mb Ma-Mb (18)
In very short hydrogen bonds (such as SSHBs or LBHBs) we may similarly distinguish between “homogeneous” and “heterogeneous” hydrogen bonds. In LBHBs a low barrier could presumably be associated with a “homogeneous” hydrogen bond in which the proton oscillates between the A and B atoms in a flat potential well.
Such spin- (or direction) conserving constraints for barrierless reactions are also present for energy transfer reactions in pigments. Förster’s expression28 for the energy transfer rate from donor to acceptor is dependent on the transition dipole moments of the donor and acceptor states. When the environments of the M atoms are differentsay, with different coordination numbers or with different orientation of the O-H dipolethe M atoms in different valence states may become distinguishable (heterogenous mixed valence). Taking this analogy between electron-transfer and proton-transfer reactions further it would seem that linear O…H-O intermolecular hydrogen bond complexes or intramolecular O…H-O complexes in which the oxygen positions are fixed are more suitable for low-barrier processes. In real molecules of biological importance the framework of the constituent atoms of the molecule could act as scaffolding that forces the A and B atoms in the AHB hydrogen bond complex to take up fixed positions and favour a low-barrier H exchange. The actual shortness of the hydrogen bond does not seem to be an important constraint. It is instead more likely that a large value of N around the hydrogen bond complex in the biological molecules reduces the O…O distance without necessarily correlating it to low barriers. In catalytic aspartases the active sites involving inner carboxylate oxygen atoms have hydrogen bond lengths between 260-287 pm. Warshel and Papazyan29/35 suggested from energy considerations show that low-barrier hydrogen bonds need not offer a catalytic advantage over ordinary hydrogen bonds.
In O…H-N bonds (prevalent in biological systems) the hydrogen atom is chemically bound to the less electronegative nitrogen atom so that the barrier to the H-O…N state from the O…H-N state may be less than the barrier to the O…H-N state from the O-H…N state. This makes the O-H…N hydrogen bond less likely to be a low-barrier hydrogen bond than the usual O…H-N bond. Steiner et al30/20 have shown from neutron diffractions studies that in short heteroatomic O-H…N with O…N distance being 258.8 pm the hydrogen atom position is sharply defined. However, the LBHB is more likely when the AHB hydrogen bond involves homo-nuclear atoms.
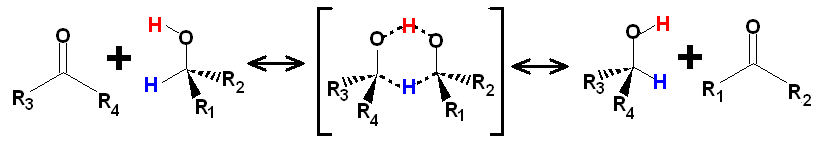
The role of LBHBs in other reactions could possibly have more potential than has been realized so far. For example, the well-known catalytic conversion of ketones to alcohols in the Meerwin-Ponndorf-Oppenauer-Verley reduction is now known to take place31, 32/23, 24 non-catalytically in supercritical isopropanol (at 300oC). From mechanistic investigation31/23 of this reaction a six-membered intermediate of this reaction features a O…H…O linkage (see above) which may be presumed to have the same features as a LBHB.
6. Conclusions
As discussed elsewhere6-8 molecular tensegrity principles are applicable to molecules in a stationary state when the = 0 condition is expected to hold25. Because of this it is sufficient to obtain atomic sizes with this condition. This condition is not uniquely defined by a unique set of atomic coordinates. It is, however, considered to be a (simple linear) function of a unique atom-specific size, rnZc (Table 1). These functions may then vary for different kinds of bonding and environment. Once a given stationary state is reached, the final geometry does not depend on the way (the mechanics) this geometry is reached. Instead, it depends on the constraints on the various interatomic distances. For an X-M-Y linkage, these constraints depend on a tensegrity factor, t00, that is a measure of the matching of ideal bonded M-X (or M-Y) and 1,3- non-bonded X…Y “charge-transfer” distances for a give X-M-Y linkage. Limiting values t00limN are obtained for different values of coordination number, N (= 4 or 6 in the present study). The calculated X…Y distance is then a simple function of the ration t00/t00limN for a given N and decreases with increasing N. The calculated distance is also a function of “n-polar” (ionic) or “neutral” (vdW) character of the X…Y contacts.
We have applied this model to more than 1000 A…H-B hydrogen bond complexes found in compounds of biologically important -amino acids with A…H distances less than the sum of their van der Waals sizes. We assume that the ideal A…H distance (sec 2) is the sum of charge-transfer sizes, CR0–, of A and H atoms and that the ideal H-B distance is the sum of the charge-transfer bonded distance, dBH00, of H and B atoms. The tensegrity factor for such hydrogen-bonded complexes is then defined (sec 4 eqns 12 and 13) and the expression for the A…B distance, dA—B, is obtained (eqn 14) from the sizes KCR0 (A, B) of the A and B atoms with K = 1 describing “n-polar” (or ionic) sizes and K = 1.125 describing “neutral” (or what may be called vdW) sizes10. The possible A…B distances are tabulated in Table 2. The comparisons of calculated distances with experimental distances are expressed as histograms of their ratios, RA—B, in various figures (Figs 4 and 6).
The O…H-O and O…H-N complexes have histograms of A…B distances that have their maximum corresponding to “n-polar” (K = 1) sizes for N = 4, while others have the maximum of their histograms close to the longer “neutral” (K = 1.125) sizes. The very short A…B distances are found mainly in O…H-O complexes with the maximum in their histograms corresponding to that calculated from eqn 14 with N = 6 (see fig 5). The inter-dependence of A…H and H-B distances with those of the A…B distances is found to be consistent with the constraints that are implicit when regarding the A…H-B hydrogen bond complex as tensegrity structures (Fig 7 and 8). Finally, we discuss the significance of low-barrier hydrogen bond complexes (LBHBs) and short hydrogen bond distances in terms of our analysis in section.
Acknowledgment.
Much of the ideas leading to this work has emerged from my work carried out with a now expired Central Scientific and Industrial Research emeritus fund from the government of India. He is thankful to the National Chemical Laboratory for desk space during this grant. He acknowledges valuable discussions on the hydrogen bond with Professor A. Nangia and Professor G. R. Desiraju of the School of Chemical Sciences at the University of Hyderabad.
References
1/36. A. D. Buckingham and P. W. Fowler, Can. J. Chem. 63, 2018 (1985).
2/2. F. Fontaine-Vive, M. R. Johnson, G. J. Kearley, J. A. Cowan, J. A. K. Howard, S. F. Parker, J. Chem. Phys., 124 (2006) 234503
3/52. W.M. Latimer, and W.H. Rodebush, J. Am. Chem. Soc., 42 (1920) 1419
4/53. W.M. Latimer, Chem. Rev., 44 (1949) 59
5/54. T.S. Koritsanszky and P. Coppens, Chem. Rev., 101 (2001) 1583
6/40. P. Ganguly, Curr. Sci. 90 1251 (2006)
7/41. P. Ganguly, Curr. Sci. 91 1505 (2006)
8. P. Ganguly, in preparation (submitted to J. Mol. Structure)
9/38. P. Ganguly (2008) J. Phys. B At. Mol. Opt. Phys. 41, 105002
10/39. P. Ganguly and G. R. Desiraju, (2008) Chemistry Asian J. 5, 868
11/37. P. Ganguly (2008) to be published
12/3. T. Steiner, Angew. Chem. Int. Ed., 41 (2002) 48-76
13/5. G. A. Jeffrey and W. Saenger, Hydrogen Bonding in Biological Structures, Springer Verlag, Berlin, 1991, p. 29.
14/6. G. R. Desiraju and T. Steiner, The Weak Hydrogen Bond: In Structural Chemistry and Biology, Oxford University Press, Oxford, 1999.
15/8. Gerlt, J. A.; Kreevoy, M. M.; Cleland, W. W.; Frey, P. A. Chem. Biol. 1977, 4, 259.
16/9. Cleland, W. W.; Kreevoy, M. M. Science 1994, 264, 1927.
17/18. W. W. Cleland, P. A. Frey, and J. A. Gerlt, J. Biolog. Chem., 273 (1998) 25529–25532,
18/48. P. Ganguly, J. Am. Chem. Soc. 117 (1995) 2655.
19/49. P. Ganguly, J. Am. Chem. Soc. 117 (1995) 1776.
20/55. P. Ganguly, unpublished results ( see http://www.materials-
chemistry.com/bond_orderJPCI.doc)
21/56. D.R. Lide ed CRC Handbook of Chemistry and Physics Internet Version 2007, (87th Edition), Taylor and Francis, Boca Raton, FL, 2007, Section 9
22/17. A. Das, V. Prashar, S. Mahale, L. Serre, J.-L. Ferrer, and M. V. Hosur, Proc. Natnl. Acad. Sci., 103 (2006) 18464-469
23/60. N. Srinivasan, B. Sridhar, R. K. Rajaram, Acta Cryst. E 57 (2001)0888
24/59. J. Marin, C. Didierjean, A. Aubry, J.-R. Casimir, J.-P. Briand and G. Guichard J. Org. Chem. 69 (2004) 130
25/61. P. Ganguly, J. Phys. Chem. A 104 (2000) 8432
26. C. Zener, Phys. Rev., 1951, 82, 403
27. See Y. Tokura, Ed., Colossal Magnetoreisitive Oxides, (Gordon and Breach Science, New York, 2000)
28. T. Förster, Delocalized excitation and excitation transfer. In Modern quantum chemistry, part III, Sinanoslu O, editor. New York: Academic Press; 1965. p. 93.
29/35. Warshel, A., and Papazyan, A. (1996) Proc. Natl. Acad. Sci. U. S. A. 93,
30/20. T Steiner, C. C. Wilson and I. Majerz, Chem. Commun., 2000, 1231–1232
31/23. T. Kamitanaka, T. Matsuda and T. Harada, Tetrahedron, 63 (2007) 1429
32/24. L. Sominsky, E. Rozental, H. Gottlieb, A. Gedanken and S. Hoz, J. Org. Chem., 69 (2004) 1492-1496
<d HB00> = 2aH
dBH00cos(HBA) = 2aH
dABO = <dAH>AO + 92 (R = 0.924 SD = 12.5) 63 + 29
dABO = 0.973<dAH>AO + 97 (R = 0.924 SD = 12.5)
dABN = <dAH>AN + 97 (R = 0.867 SD = 13) 71 + 26
dABN = 0.796<dAH>AN + 137 (R = 0.867 SD = 12)
dABC = <dAH>AC + 110 (R = 0.906 SD = 12) 83 + 27
dABC = 0.735 <dAH>AN + 171 (R = 0.906 SD = 10)
Intercept = 4.11rnZc(B) + 30 pm